

How to better understanding Variants of Uncertain Significance in epilepsy and help find new therapeutic approaches
Did you know that 1 in 26 people will experience epilepsy at some point in their lifetime?
This statistic is very high, and therefore it is likely many of you have experienced epilepsy or know someone who has. My experience was with my son. When my boy Alemeyahu was just about to turn 2 years old, I had him outside on a beautiful sunny day. Bouncing on my knee, giggling away when suddenly his head tilted back and his eyes rolled up in his head. He went limp and stopped breathing.
Panicking I laid him down on the ground and yelled for help while I lifted his chin and started mouth to mouth.
Diagnostic Rate in Epilepsy

Photo credit: eztv.io
With the high rate of epilepsy in the population there is a bit of a puzzle out there. How is it that the diagnostic rate of genome sequencing is so low? A recent retrospective study of 8565 epilepsy patients was done for patients who underwent genetic sequencing diagnostic test. The ability to diagnose a genetic cause to their epilepsy was only 15%. What of the remaining 85% – much of that remains dark matter of uncertainty.
Dark Matter Problem – what is the cause?
What is the major driver to low diagnostic rate with a DNA sequencing test? When we look at variants occurring in the genome, there are tens of thousands of variants in the coding sequences for important genes in each person’s genome. Many of these genetic differences are benign, but some variations may be pathogenic drivers of disease.
There are over 100 genes that are involved in causing epilepsy. So the clinician/geneticist has their work cut out for them in trying to decide which variants they can dismiss and which one should be of concern. Many of the variants are challenging to dismiss, which can leave us with a bit of concern.
80,000,000 Variant Possibilities
There are roughly 80 million places to pick up an amino acid change (average gene size: 600 bp x number of disease genes: 7000 x amino acids: 20). Granted maybe only 10% may actually be in places of concern, but that is still a large number and we have only just scratched the surface. Clinvar database can be mined for number of variants known to be pathogenic or benign. As of December 2018, the number of pathogenic variants (P + LP) is over 115,000. The number of benign (B + LB) is over 168,000. On the 80 million, that is only 0.35% that are known to either pathogenic or benign, the remaining 99+% is a form of genomic dark matter!
Is low pathogenicity of genome to be expected?
When we look at variant calls for highly studied genes the answer is no. ClinVar data was examine for the top 20 genes with the most number of submitters supplying variant data. On average 51 groups per gene submitted sets of variants. Total variant assessments submitted were 72,471 at an average of nearly 3000 calls per gene. Taking every amino acid position in this top 20 we have a theoretical variant space of 1, 411,833. As a result, 5% of the theoretical space has been covered. Yet the amount of pathogenicity is rather steady (in fact it has gone up a bit!).
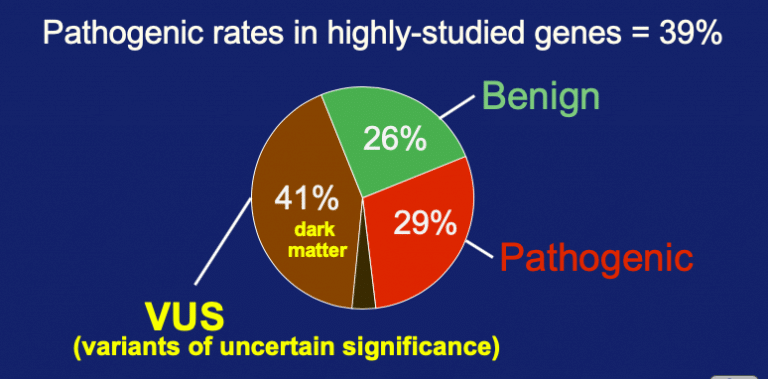
In the 5% coverage, there were 34% as pathogenic + likely-pathogenic. A nearly equal number were benign + likely-benign. And in what may seem to be a bit of a surprise, the variants of uncertain significance are still high at 40% of the entries. So pathogenicity extended is trending at about a third of examined rare variants and VUS is trending at close to 40%.
What are the data trends over time?
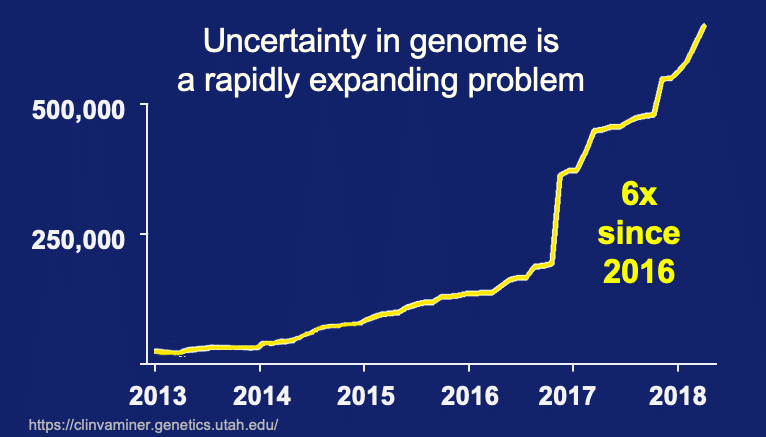
Couple the large problem of Variants of Uncertain Significance with data trends and we have an alarming phenomenon looming ahead of us. As previously posted, Clinvar data was extracted for the rate of new submissions since 2013. We see the number of new submission is starting to accelerate in just the last few years. To sum this up – an avalanche of uncertainty is coming and we are ill prepared to deal with it.
It is just a febrile seizure episode.
My son Alemeyahu was still not breathing. After I put about 5 breaths of air into his lungs and I checked his pulse – thankfully his heart was still beating, but he was still not breathing! I put more air into his lungs and finally, he breathed a weak breath. Then another and another – he was finally breathing again.
I called the paramedics and by the time they got there, Alemeyahu was back to being a normal 2 year old – trying to crawl over the downstairs barricade, trying to put thumb size rocks in his mouth, you know, the usual 2 year old stuff!
We got him to the hospital where they ran all kinds of tests but they couldn’t find anything wrong. They diagnosed it as a Febrile Seizure Episode and said “Bring him back if it repeats.” Well, ever since, there have been no repeat episodes, but for others, people with chronic epilepsy, they sometimes have to deal with repeats on a daily basis. Sometimes even hour to hour, and many of the current epilepsy drugs don’t work well for 1/3rd of all epilepsy cases. So, we must find a way to help people with epilepsy achieve better control of their seizures symptoms. Humanized animal models may be one key system to help us understand variant biology and find new therapeutic options in epilepsy.
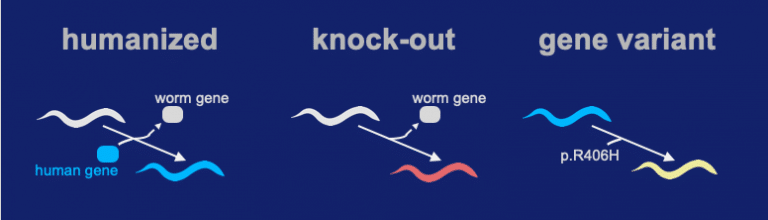
Our team successfully inserted the human coding sequence of STXBP1 into the native locus of a C. elegans ortholog gene. We call the technique a “Gene-Swap” because the worms version of the gene is replaced with a human coding sequence. The Gene-Swapped STXBP1 sequence functioned and gave a high level of rescue activity (the “Humanized” strain). A knock-out was made by precisely deleting the unc-18 locus (ortholog of STXBP1). In the process, all coding sequence of unc-18 is removed and a full loss-of-function deletion allele is generated. In the gene variant we insert the p.Arg406His into the humanized locus.
The gene-swap humanized strain for expressing hSTXBP1 shows a significant level of activity (see video above). In contrast, the knock-out shows very little activity. For the R406H genomic variant, its activity is somewhere in between the “humanized” and “knock-out” strain.
Behavior quantified using three assays
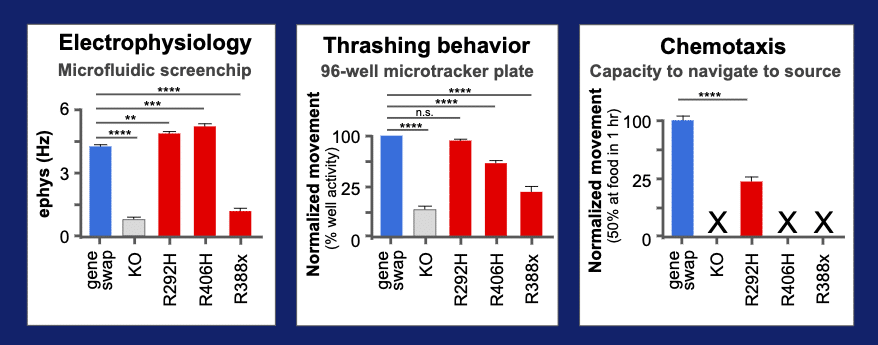
R406H’s deviant behavior was quantified in a microfluidics system that measures an EEG-like electrical activity of the animal. We added two other assays on this variant – a wMicroTracker test for thrashing in liquid and and a chemotaxis test for speed to navigate to a food source. We also added two other reference alleles that are also known to be pathogenic variants of STXBP1 (R292H and R388X). The three variants in red can be compared to the wild type “gene-swap” for the humanized line in blue. In the elecrophysiology assays, we can see that two variants, R292H and R406H, have increased rate of neurotransmission. In contrast, the R388X allele has a lower rate of transmission. Adding in the thrashing assays for movement in liquid, we see that the R292H has lost its deviant behavior, while the R406H and R338X have retained a detectable level of altered function. A final assays was performed that uses a sensitive chemotaxis behavioral assay. This assay measure the ability of animal to reach a food source in 1 hr. All variants show altered function and only the R292H allele shows residual level of activity, so we can see that a series of assays provides a measure of pathogenic variant biology and gives a glimpse into the mechanism of disease.

Speed is an important aspect to measuring variant biology. Our team did an internal contest. We wanted to see how fast we could install a variant and measure activity. In the end, we were able to do it and provide an assessment in just under 10 days.
The variant profiling system has three key values:
- First, because it uses a gene-swapped animal expressing a human gene, any variant can be installed and profiled for functional defects.
- Second, a series of assays can be deployed to detect the nuanced differences in variant pathogenicity.
- Third, the humanized animal models can be used in high-throughput screens as discovery systems for finding personalized therapeutics.

